Question
« Une entreprise du secteur des technologies de la santé a lancé un rappel mondial de ses appareils respiratoires. Ce qui est inquiétant, c’est que ce rappel a déjà été lancé en juin 2021. Mais ici, au Luxembourg, ce n’est que fin octobre 2022 que l’on en a eu connaissance. Et ce, après qu’un patient concerné ait attiré l’attention sur ce rappel.
Dans ce contexte, j’aimerais poser les questions suivantes à Madame la Ministre de la Santé :
- Quel département est responsable de la communication des rappels de dispositifs médicaux ? Comment les procédures de communication devraient-elles se dérouler ?
- Pourquoi n’a-t-on pas réagi à temps à ce rappel d’appareils respiratoires ?
- Madame la ministre, estime-t-elle que les procédures de communication devraient être adaptées afin d’éviter un tel cas à l’avenir ? Dans la négative, pourquoi ?
- Combien de fois y a-t-il eu des rappels de dispositifs médicaux au cours des dix dernières années ? Combien de temps a-t-il fallu pour que toutes les personnes concernées aient été informées ?
- Madame la Ministre, ne juge-t-elle pas utile de finaliser le projet de loi sur l’Agence Luxembourgeoise des Médicaments et Produits de Santé (ALMPS) dès que possible ? Où en sont actuellement les travaux relatifs au projet de loi ? »
Réponse
Quel département est responsable de la communication des rappels de dispositifs médicaux ? Comment les procédures de communication devraient-elles se dérouler?
L’article 8 du règlement grand-ducal modifié du 11 août 1996 relatif aux dispositifs médicaux indique que la Direction de la santé est en charge de recevoir, recenser et évaluer toute donnée communiquée en lien avec des incidents. L’article 4(5) de la loi modifiée du 21 novembre 1980 portant organisation de la Direction de la santé stipule que « la division de la pharmacie et des médicaments (DPM), au sein de la Direction de la santé, a compétence pour toutes les questions relatives aux dispositifs médicaux ».
L’article 89(8) du Règlement européen 2017/745 précise que c’est le fabricant qui doit procéder au rappel/retrait du dispositif non-conforme, tout en communiquant en parallèle les informations pertinentes à l’autorité compétente.
Pourquoi n’a-t-on pas réagi à temps à ce rappel d’appareils respiratoires ?
La procédure de rappel initiée par le fabricant a été suivie au fur et à mesure de l’information par le fabricant des distributeurs luxembourgeois et du remplacement des appareils. Il convient de préciser que le dispositif médical en cause a été vendu sous différents noms de marque et par différents distributeurs au Luxembourg. Il est important de noter qu’à ce jour, aucun incident concernant ces appareils n’a été rapporté au Luxembourg. Il faut aussi insister sur le fait qu’il a été recommandé par le fabricant de ne pas arrêter le traitement avant d’avoir consulté son médecin, quel que soit le type d’appareil utilisé.
Il faut cependant remarquer qu’il y a eu de nombreux retards dans la mise en œuvre de cette action corrective car Philips Respironics n’a pas été en mesure de réparer ou de remplacer tous les appareils concernés en raison de l’ampleur énorme du rappel, concernant plusieurs pays et des millions de patients. A cet égard, de nombreux pays européens ont fait part de leurs inquiétudes et tenté de faire pression sur le constructeur dans une action coordonnée par l’autorité française compétente (Agence Nationale de Sécurité du Médicament et des Produits de Santé –ANSM), cependant sans résultat notable.
Madame la ministre, estime-t-elle que les procédures de communication devraient être adaptées afin d’éviter un tel cas à l’avenir ? Dans la négative, pourquoi ?
Consciente des améliorations nécessaires en matériovigilance, l’Union européenne a légiféré et dans le cadre général de la traçabilité des dispositifs médicaux, des améliorations sont prévues par le Règlement européen 2017/745 grâce à la mise en place d’un identifiant unique (IUD-ID) sur l’étiquetage pour les dispositifs médicaux de classe III (c.à.d. les dispositifs à risque élevé) tombant sous les dispositions dudit règlement.
En application de ce règlement, la mise en place d’un enregistrement des distributeurs mettant des dispositifs médicaux à disposition sur le territoire luxembourgeois, ainsi que des dispositifs médicaux mis à disposition sur le territoire luxembourgeois, est en préparation.
Afin d’améliorer encore l’efficacité des missions incombant à l’autorité compétente pour l’enregistrement, ainsi qu’à l’autorité de surveillance du marché, en charge des dispositifs médicaux et des dispositifs médicaux de diagnostic in vitro, une demande d’analyse des besoins d’une base de données liée aux tâches nationales et connectée à EUDAMED est en cours.
La page internet de cette base de données pourrait être conçue comme une porte d’entrée unique pour les professionnels de la santé et les fabricants pour la notification d’incidents, et aussi permettre la publication des notices de sécurité relatives aux dispositifs médicaux défectueux.
Les procédures de communication pourraient donc être améliorées, mais chaque chaîne d’approvisionnement nécessite des adaptations spécifiques suivant le type de dispositif ainsi que son niveau de risque.
Combien de fois y a-t-il eu des rappels de dispositifs médicaux au cours des dix dernières années ? Combien de temps a-t-il fallu pour que toutes les personnes concernées aient été informées ?
Le tableau ci-dessous reprend le nombre de notifications par année, plusieurs notifications concernant parfois un seul produit étant donné qu’un suivi d’information de l’action en cours doit être assuré par le fabricant. Ces chiffres ne représentent à minima les échanges effectués entre la DPM et les producteurs, seuls les notifications formelles des firmes y sont reflétées. En effet, les échanges de suivi effectués par la DPM varient en fonction des produits et des problèmes rencontrés. La compilation de ces statistiques n’ayant débuté qu’en août 2016, le tableau ne présente que les données annuelles à partir de l’année 2017 (les chiffres de 2022 couvrent la période allant jusqu’au mois d’octobre).
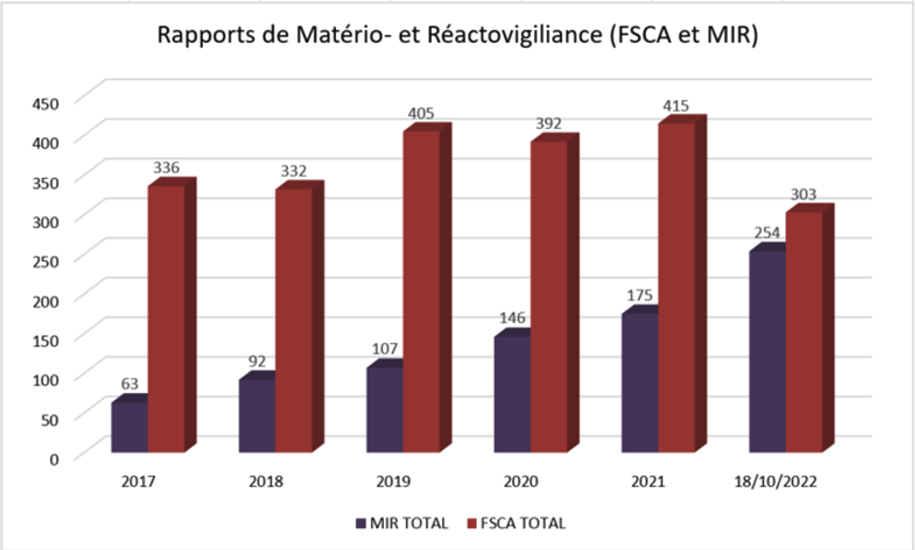
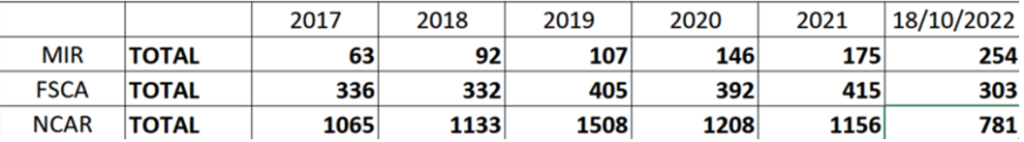
MIR – Manufacturer Incident Report
FSCA – Field Safety Corrective Action
NCAR – National Competent Authority Report
Madame la Ministre, ne juge-t-elle pas utile de finaliser le projet de loi sur l’Agence Luxembourgeoise des Médicaments et Produits de Santé (ALMPS) dès que possible ? Où en sont actuellement les travaux relatifs au projet de loi ? Les travaux relatifs à l’avant-projet d’amendements gouvernementaux au projet de loi 7523 visant la création d’une agence nationale des médicaments et des produits de santé sont actuellement en cours et devraient être soumis au Conseil de Gouvernement avant la fin de l’année.